Software
Many of our projects are available via the Novembre Lab Github Page. Here are some of our software projects:
feems (fast-EEMS)
A statistical method for inferring and visualizing gene-flow in spatial population genetic data, developed by PhD student Joe Marcus
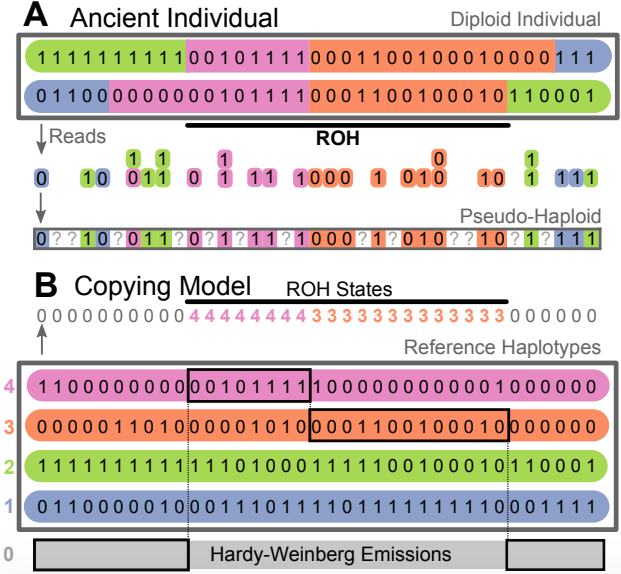
hapROH
Identify runs of homozygosity (ROH) in low coverage ancient human DNA data using modern haplotype reference panels, developed by former postdoc Harald Ringbauer.
geovar
A package to visualize joint allele frequency spectra from population genetic samples, developed by former PhD student Arjun Biddanda.
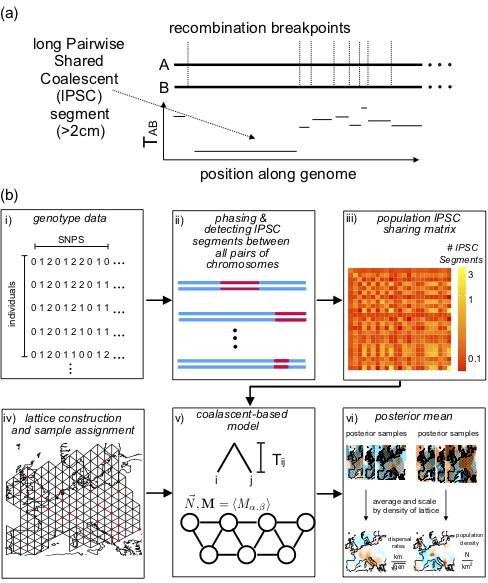
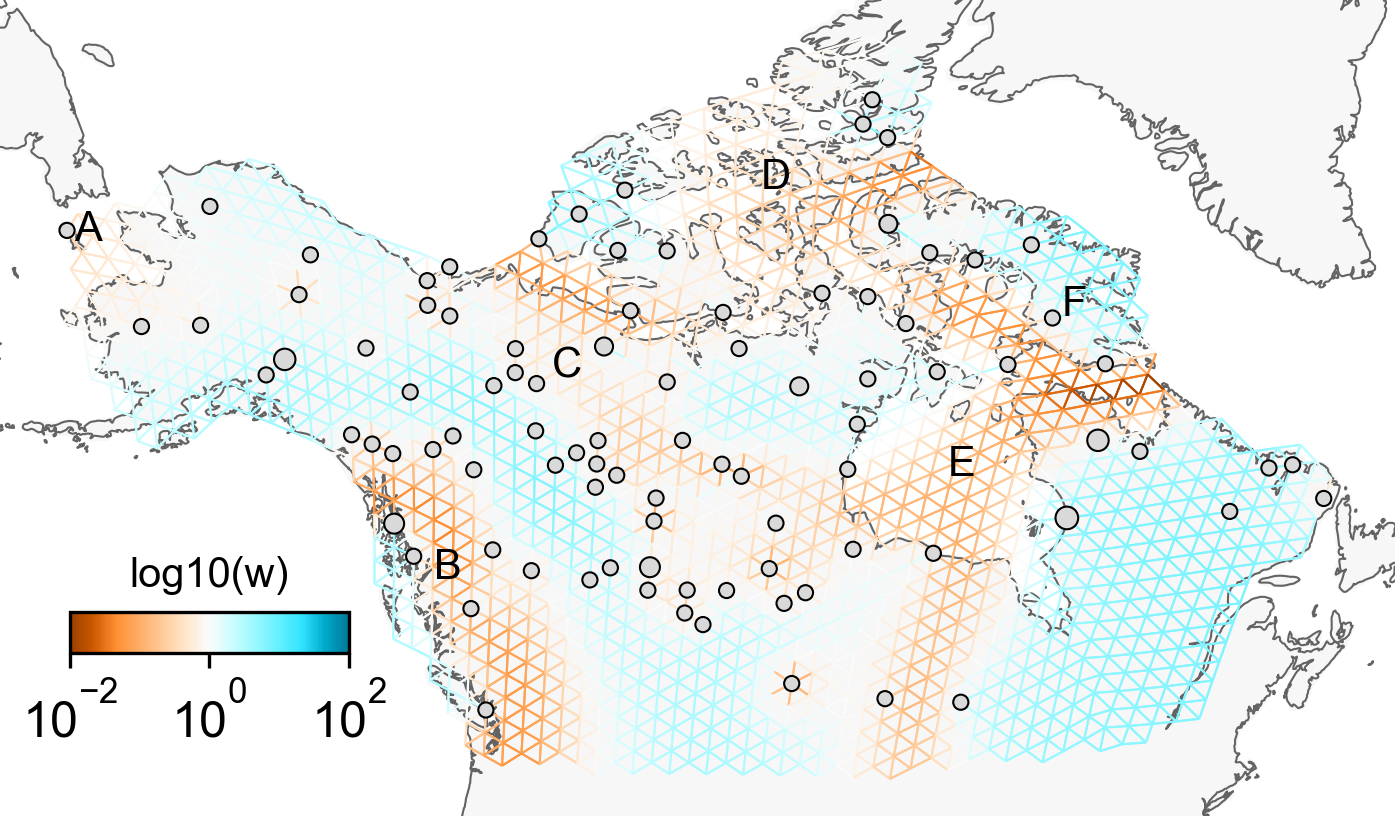
MAPS
A method for estimating dispersal rates and population densities based on the number of shared long Pairwise Coalescent Segments (lPSC, aka “IBD tracts”), developed by former PhD student Hussein Al-asadi.
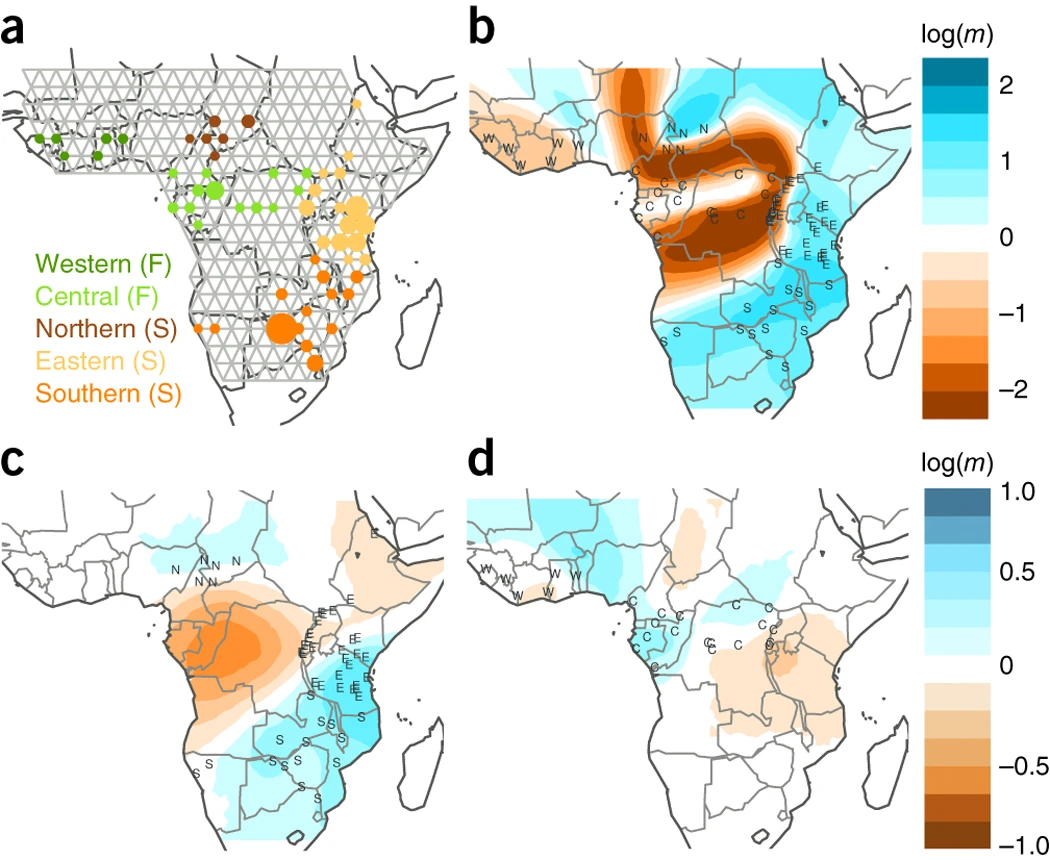
EEMS
A method for visualizing population structure in spatial data using “effective migration” surfaces, developed by Desi Petkova
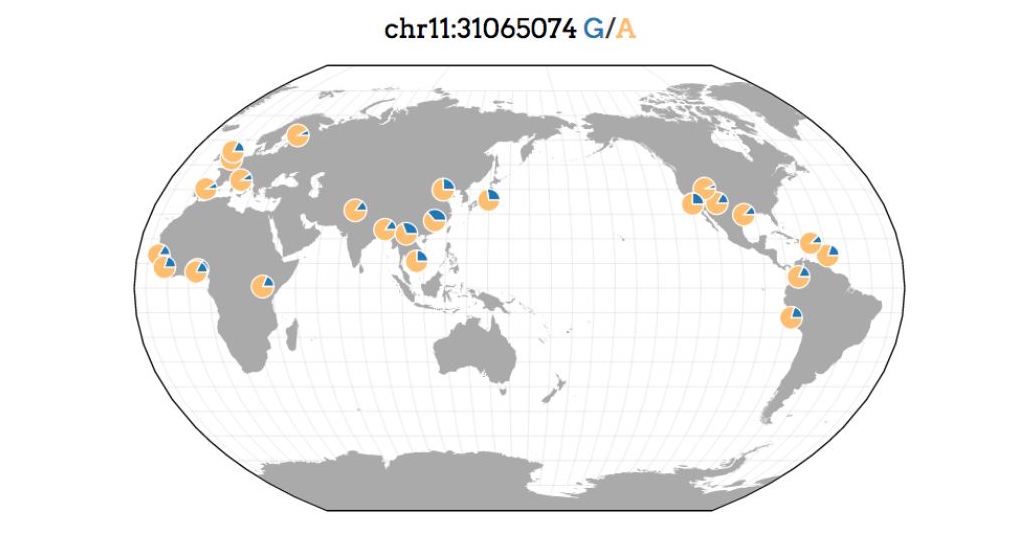
Geography of Genetic Variants Browser
A web-based tool for visualizing the geographic distribution of variants in several published datasets.
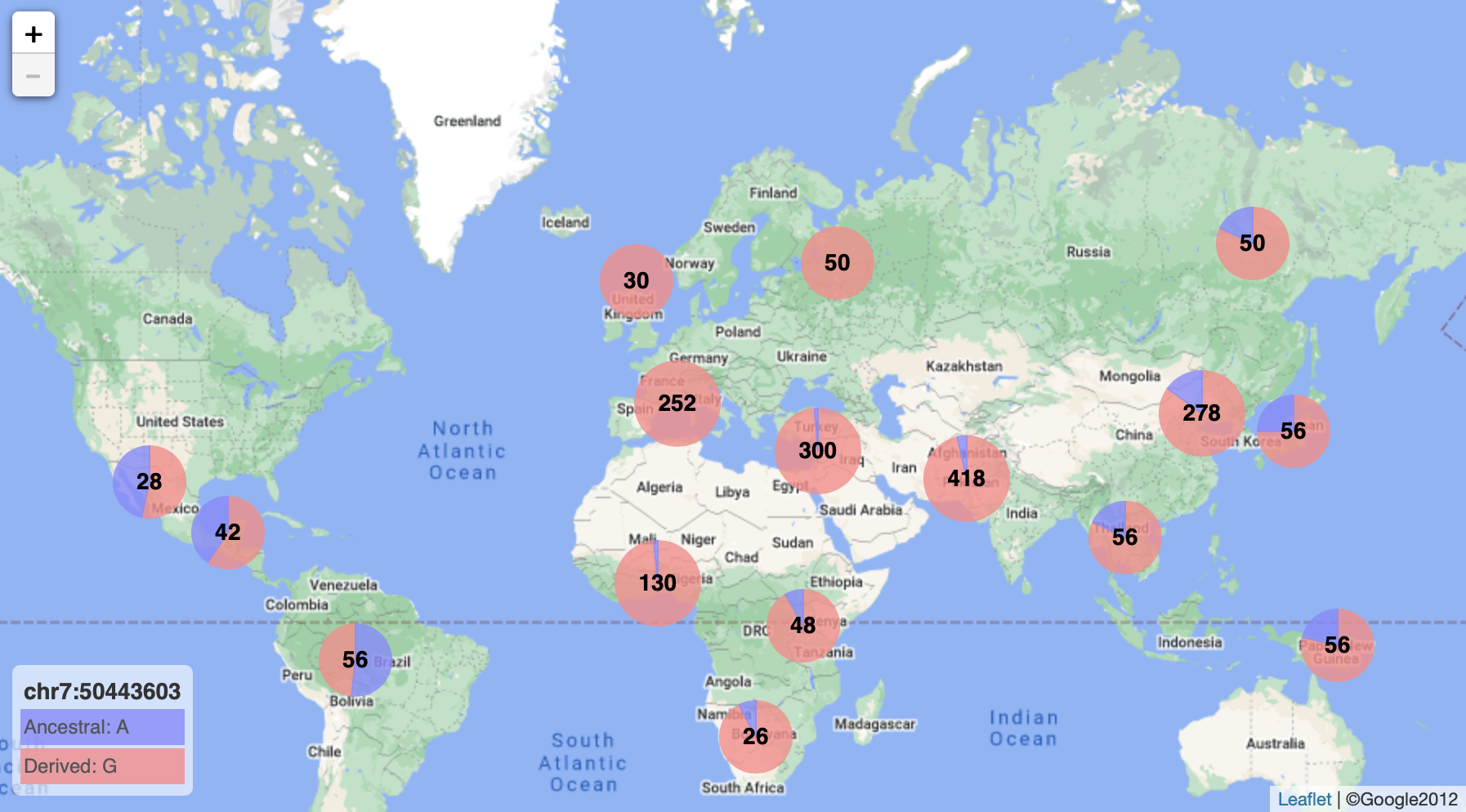
RGGV
An R package that for plotting of allele frequencies on geographic maps, developed by David Witonsky.
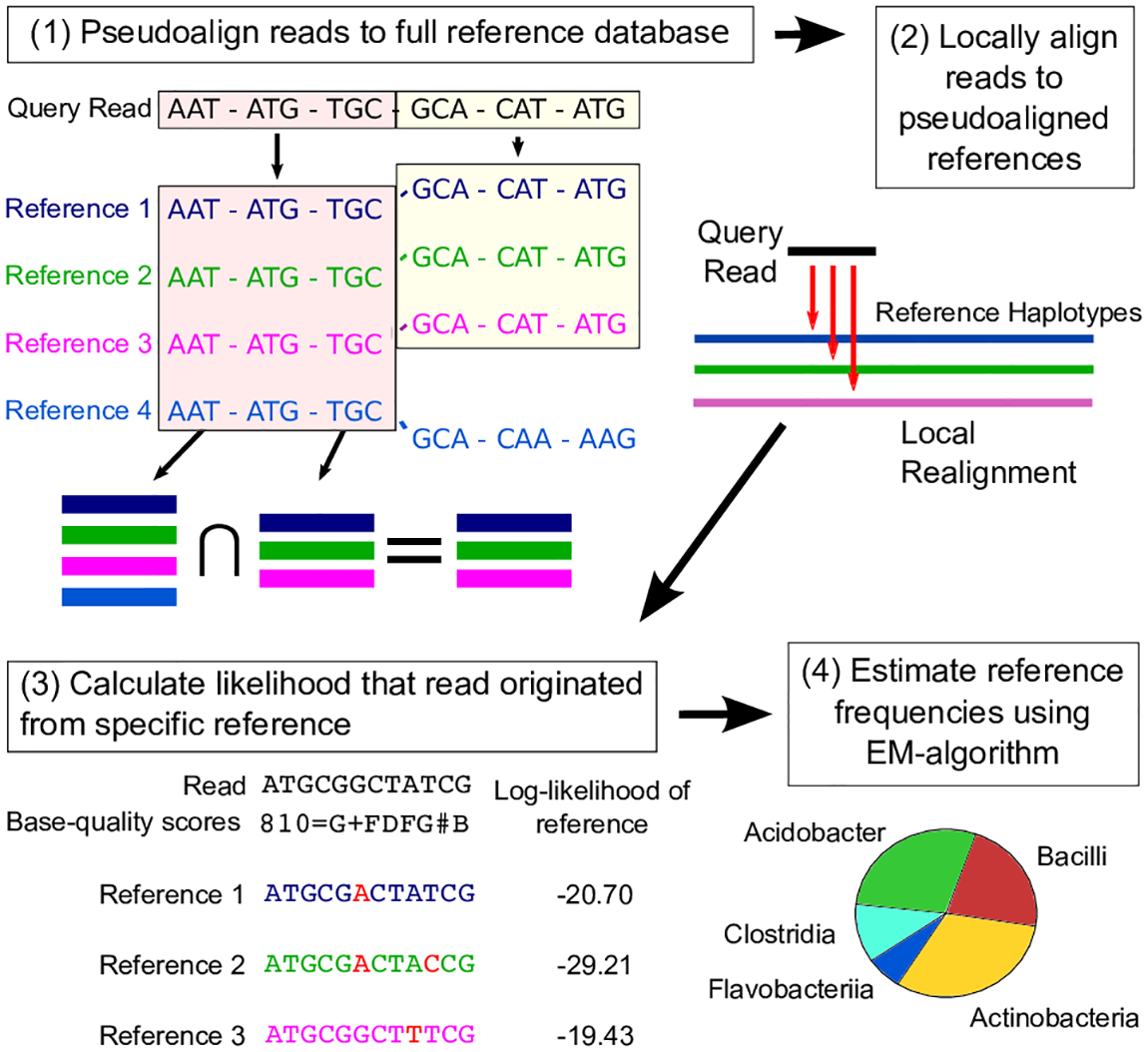
karp
A method for k-mer based analysis of read pools using psuedo-alignment and base quality information, developed by former postdoc Mark Reppell.
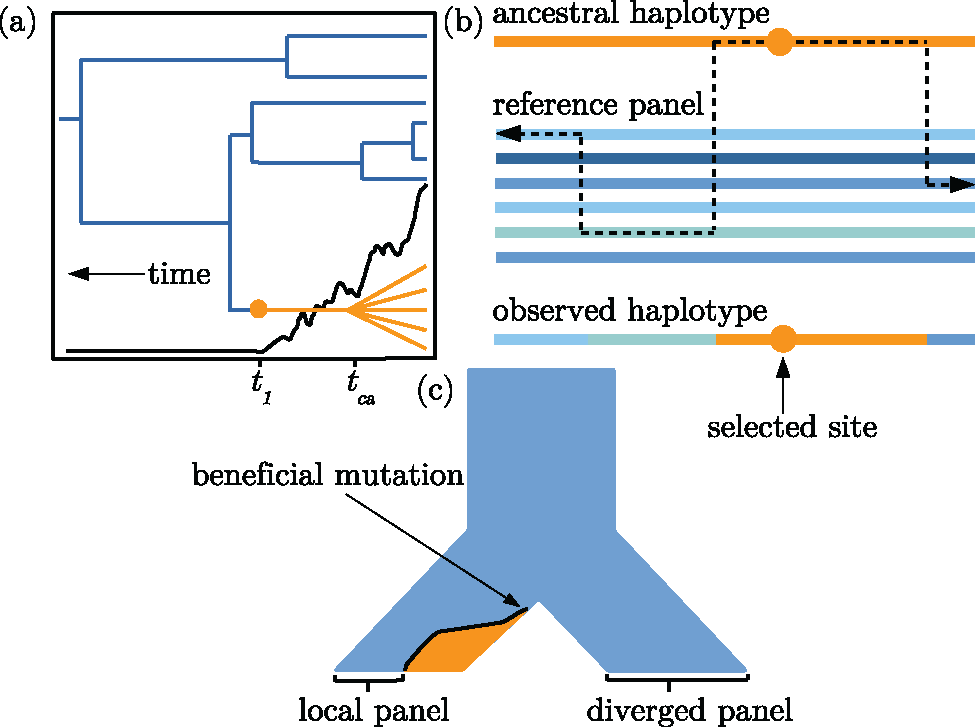
starTmrca
A method to estimate the timing of selective sweeps from haplotype data, developed by PhD Student Joel Smith.
forqs (Forward-in-time simulation of Recombination, Quantitative traits, and Selection)
A flexible forward-in-time simulator, developed by PhD student Darren Kessner.
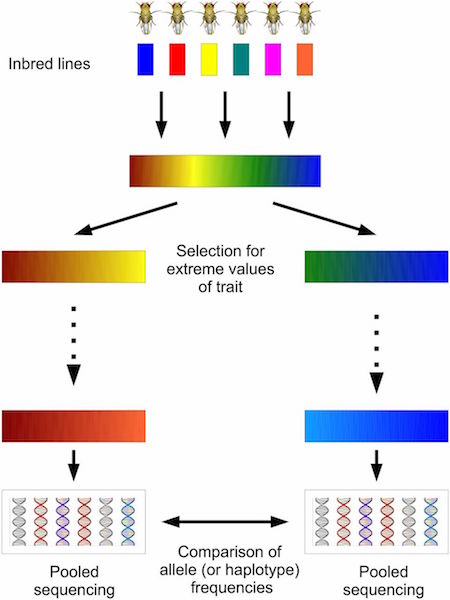
harp (Haplotype Analysis of Reads in Pools)
Maximum likelihood estimation of frequencies of known haplotypes from pooled sequence data, developed by PhD student Darren Kessner.
RASPberry (Recombination via Ancestry Switch Probabilities)
Software for recombination rate inference using ancestry switch points in admixed individuals, developed by former postdoc Daniel Wegmann.
ADMIXTURE
Software for ancestry inference in population genetic data developed by former UCLA PhD student David Alexander.
isoscatR
An R-package for spatial assignment using genetic and isotopic data, developed by former PhD student Colin Rundel, and based in part on the SCAT software from Matthew Stephens.
raddle, Rare Allele Distance of DispersaL Estimation
The Raddle software for Rare-Allele-based Distance of Dispersal Estimation, based on Novembre & Slatkin (2009)
MDBlocks
A program written with Eric Anderson (when we were both at Berkeley) that partitions a chromosomal segment into haplotype blocks using biallelic, phased haplotype data.
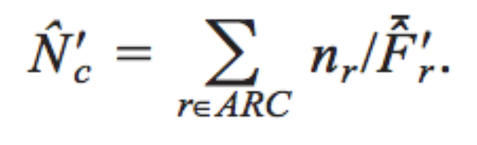
ENCprime
A program that calculates a codon usage bias summary statistic, Nc’. It is based on the effective number of codons statistic Nc (or ENC) developed by Frank Wright, but improves upon it by accounting for background nucleotide composition.
SIMCOAL
A program that uses a coalescent framework to simulate the evolution of multiple haploid populations with complex demographies . Developed by Laurent Excoffier and Stefan Schnieder with some contributions from JN. NOTE- A new version, fastSIMCOAL2, is now available.
Data Resources
Resources related to Novembre et al 2008: Several files that aid in recreating the main figure from our paper:“Genes Mirror Geography in Europe” (Nature 956:98-101).
African-American/African-Caribbean recombination maps: Recombination maps inferred from a sample of 2565 African Americans and 299 African Caribbeans. From our paper: Wegmann et al (2011) Nature Genetics 43:847-53.
Resources for Freedman et al 2014: Fastq files are available via SRA. Auxiliary files such as vcf/bams are available via an sftp server. Please email – jnovembre at uchicago dot edu – for user and password.